







Shared Multi-Omic Data for Advancing Neuroendocrine Neoplasm Research
NETRF-funded researchers have recently reported in the journal GigaScience the first multi-omic dataset of patient-derived
NETRF-funded researchers have recently reported in the journal GigaScience the first multi-omic dataset of patient-derived
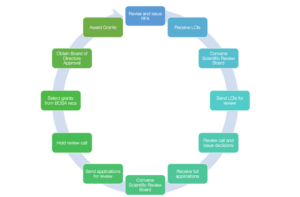
At NETRF, our mission is to fund research that holds promise for advancing the understanding

Recent advancements have broadened the spectrum of treatments for advanced NETs, encompassing a range of